糖原贮积病Ⅱ型
糖原贮积病Ⅱ型是一种罕见的进展性溶酶体贮积病,由位于第17号染色体上的酸性α-葡糖昔酶(GAA)基因突变所致,呈常染色体隐性遗传。
疾病知识
症状
糖原贮积病Ⅱ型临床分型依据发病年龄分为
婴儿型和晚发型。发病年龄与a葡糖苷酶缺乏程度有关,葡糖苷酶降低程度越严重,发病年龄越早,
病情进展越快。
1.婴儿型于1岁内发病,主要累及骨骼肌和
心肌,表现为“软婴儿”,四肢肌力、肌张力下降。典型患儿于新生儿期至出生后3个月内发病,呈进行
性肌无力、运动发育迟缓、喂养困难、吞咽困难,查
体可见心脏扩大、心肌肥厚、肝肿大、舌体胖大;呼
吸肌受累导致通气性呼吸衰竭,反复肺炎,气管插
管后易产生呼吸机依赖。肥厚型心肌病早期表现
为心室肌和室间隔肥厚,心室流出道梗阻,随后出
现扩张型心肌病,亦可累及心脏传导系统,增加室
性心律失常和猝死风险。患儿常于1岁左右死于循
环、呼吸衰竭。
2.晚发型于1岁后发病,亦可至成年期发病,
主要累及躯干和四肢近端骨骼肌和呼吸肌。依据发病年龄又分为儿童型和成年型。儿童型主要表
现为运动发育迟缓、四肢无力,Gower征阳性;成年
型以缓慢进展的肢体近端肌无力为主,下肢较上肢明显,腰肌无力或屈髋无力可以是最早表现。呼吸肌无力十分常见,表现为咳嗽无力、呼吸困难,严重时需呼吸机辅助通气。少数患者以呼吸肌无力发
病。甚至可因呼吸障碍首诊于呼吸科。约有74%
的患者存在呼吸功能下降,38%表现为明显的膈肌无力。呼吸肌无力是导致晚发型患者死亡的重要原因。晚发型患者心肌较少受累,部分患者可有不
同程度血管病变,如胸主动脉瘤、基底动脉瘤等。
也有少数患者以血管病变作为首发表现,而后才出
现肌肉病症状。成年型患者的临床过程一般好于
婴儿型和儿童型。肢带肌无力和呼吸肌无力是成年型的主要表现,翼状肩胛、股四头肌萎缩和眼睑下垂等也十分常见。
病因
编码酸性α-葡糖苷酶(acid alpha-glucosidase,GAA)的基因——GAA基因(MIM 606800)位于17q25.3,由于GAA基因突变,溶酶体内GAA活性缺乏或显著降低,糖原不能被降解而沉积在骨骼肌、心肌和平滑肌等细胞的溶酶体内,导致溶酶体肿胀、细胞破坏及脏器功能损害,并引起一系列临床表现。根据发病年龄、受累器官和疾病进展速度,临床上将GSD Ⅱ分为婴儿型和晚发型两大类。发病率约1/40 000~1/50 000活婴,但存在种族及地区差异。2006年人重组酸性α-葡糖苷酶(rhGAA)正式应用于GSD Ⅱ治疗后,患者的预后明显改善。
检查
绝大多数患者实验室检测血清肌酸激酶(CK)
水平升高,为正常参考值的4~10倍。外周血涂片
糖原染色以镜下可见淋巴细胞胞质空泡变性,作为初筛方法。针极肌电图检查多呈肌源性损害,可出现纤颤电位、复合性重复放电、运动单位电位
(MUP)时限缩短和波幅降低等,部分患者肌电图无
特征性改变。选择近端肌肉行针极肌电图可提高阳性检出率,特别是椎旁肌和下肢近端肌肉。
针极肌电图正常者不能排除诊断。
1.肌肉活检病理特征为肌纤维呈空泡变性,
空泡大小和形态各异,糖原染色阳性,溶酶体酸性磷酸酶染色呈强阳性。
2.a-葡糖苷酶活性测定外周血淋巴细胞、组织培养(含纤维母细胞)和肌肉组织,a葡糖苷酶活性
缺乏(<1%)或显著降低(<正常参考值2%~40%),
是明确诊断糖原累积病Ⅱ型的金标准。近年来,随
着实验室检测技术的改进,外周血白细胞和干血滤纸片a-葡糖苷酶活性检测结果十分可靠,且具有方
便、快速、无创等优点,业已成为糖原贮积病Ⅱ型的
一线诊断方法。
3.基因突变分析GAA基因定位于q25.3,包含
20个外显子,编码一条含952个氨基酸的多肽,目
前已发现300余种突变类型。糖原贮积病Ⅱ型呈常
染色体隐性遗传,理论上必须2个等位基因均突变
(纯合或杂合突变)方致病。
诊断
2013-05-14国内《糖原贮积病Ⅱ型诊断及治疗专家共识》提出了糖原贮积病Ⅱ型的临床诊断路径。由于酶替代治疗正在逐渐成为可能,早期明确
诊断更显重要。尽管临床分型不同,但其诊断路径是一致的。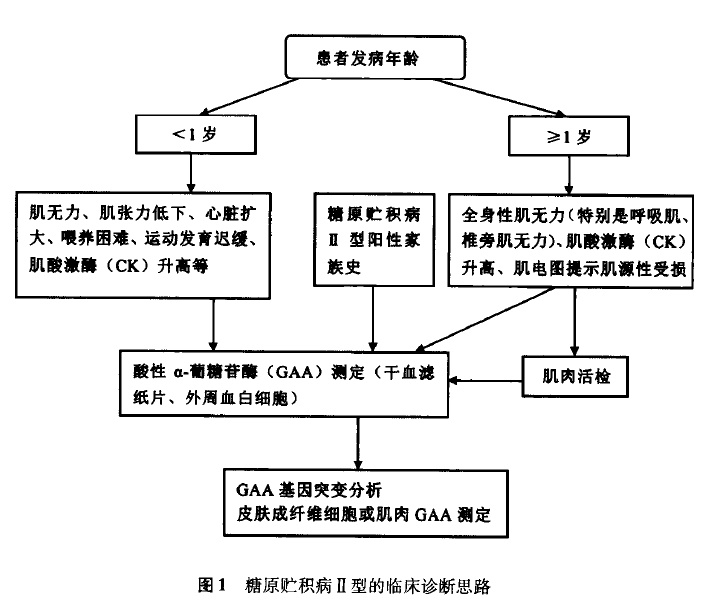
治疗
对于糖原贮积病Ⅱ型这一多系统损害性疾病,
建立多学科合作团队参与治疗管理至关重要,包括遗传代谢病专科、心脏科、呼吸科、神经科、重症医学科、理疗科、营养科和心理科等。可以居家应用
的无创性辅助通气设备有助于提高患者生活质量,
减少住院周期。酶替代治疗是该病治疗方法的重
要进步,人重组a-葡糖苷酶括Myozyme和
Lumizyme已通过美国食品与药品管理局(FDA)审
批,分别用于婴儿型和晚发型糖原贮积病Ⅱ型的治
疗。对于婴儿型患儿,明确诊断后应尽早治疗,可
显著延长生存期、改善运动功能和心脏功能;迟发
型患者出现症状与体征前,每隔6个月对肌力和肺功能进行一次评价,一旦出现症状与体征,应尽早开始酶替代治疗。人重组a-葡糖苷酶治疗前,应组织多专科进行系统评价,制定个体化治疗方案、治疗目标和随访计划。
预后
国外学者对2002—2009年
诊断与治疗的268例未接受酶替代治疗的成年型患者的临床资料进行回顾分析,患者平均诊断年龄为
38岁,诊断后平均生存27年,其中依赖轮椅或呼吸
机支持的患者5年生存率约为75%。
国内学者回顾性分析2005至2012年上海儿童医学中心确诊的16例典型性婴儿型GSDⅡ患儿首发症状年龄的中位数为3.6(2.0~6.8)个月,确诊时年龄的中位数为6.5(3.8~9.3)个月;16例患儿中14例已经死亡,死亡时年龄的中位数为9.0(4.7~18.7)个月。
预防
早期诊断,尤其是症状前诊断,早期酶替代治疗可显著改善婴儿型GSD II的预后,我国台湾地区及
部分欧美国家已经开展GSD II的新生儿筛查,作为扩大的新生儿筛查的一部分,多采用串联质谱仪法
或荧光法检测干血滤纸片GAA活性。对于筛查阳性者,建议采集外周血白细胞或淋巴细胞进行
GAA测定,如果GAA活性降低,则应同时进行遗传咨询、多学科临床评估、GAA基因突变分析以明确诊断。确诊为婴儿型GSD II者,立即开始酶替代治
疗并长期随访;诊断为晚发型者,每6~12个月随访
1次,评估临床表现,当出现肌无力和(或)呼吸功能减退或CK升高即开始酶替代治疗。我国大陆
地区尚未开展GSD 1I的新生儿筛查。
健康问答

词条标签
乏力
网友、医生言论仅代表其个人观点,不代表本站同意其说法,本站不承担由此引起的法律责任
微医提供平台支持 Copyright 2011-2017版权所有。 浙ICP备15034772号-2
